人工智能模型发现潜在药物分子的速度快一千倍
几何深度学习模型比最先进的计算模型更快、更准确,降低了药物试验失败的机会和成本。
整个已知宇宙充满了无限数量的分子。但是,这些分子中有多少部分具有潜在的类似药物的特征,可用于开发挽救生命的药物治疗?数百万?数十亿?万亿?
答案是:novemdecillion,或 1060.这个庞大的数字延长了Covid-19等快速传播疾病的药物开发过程,因为它远远超出了现有药物设计模型的计算能力。从这个角度来看,银河系大约有1亿,或108星星。
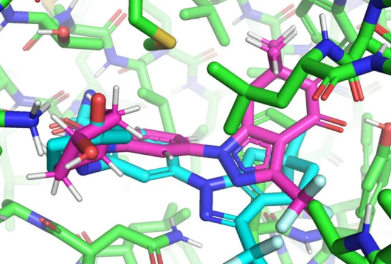
EquiBind(青色)预测可以放入蛋白质口袋(绿色)的配体。真正的构象是粉红色的。插图作者:研究人员 / MIT
在将在机器学习国际会议(ICML)上发表的一篇论文中,麻省理工学院的研究人员开发了一种名为EquiBind的几何深度学习模型,该模型比现有最快的计算分子对接模型之一QuickVina2-W快1,200倍,成功地将类似药物的分子与蛋白质结合。
EquiBind基于其前身EquiDock,EquiDock专门使用已故的Octavian-Eugen Ganea开发的技术结合两种蛋白质,该技术是最近的麻省理工学院计算机科学和人工智能实验室,安利捷健康机器学习诊所(Jameel Clinic)博士后,后者也是EquiBind论文的合著者。
在药物开发甚至发生之前,药物研究人员必须找到有前途的类似药物的分子,这些分子可以在称为药物发现的过程中正确结合或“对接”到某些蛋白质靶标上。成功与蛋白质对接后,结合药物(也称为配体)可以阻止蛋白质的功能。如果这种情况发生在细菌的必需蛋白质上,它可以杀死细菌,为人体提供保护。
然而,药物发现的过程在财务和计算上都可能代价高昂,数十亿美元投入到这一过程中,经过十多年的开发和测试,然后才获得美国食品和药物管理局的最终批准。更重要的是,90%的药物一旦在人体中进行测试就会失败,因为它们没有效果或副作用太多。制药公司弥补这些失败成本的方法之一是提高成功药物的价格。
目前寻找有前途的候选药物分子的计算过程是这样的:大多数最先进的计算模型依赖于大量的候选药物采样以及评分,排名和微调等方法,以获得配体和蛋白质之间的最佳“拟合”。
Hannes Stärk是麻省理工学院电气工程与计算机科学系的一年级研究生,也是该论文的主要作者,他将典型的配体与蛋白质结合方法比作“试图将钥匙放入具有大量钥匙孔的锁中”。典型的模型在选择最佳模型之前,会耗时地对每个“拟合”进行评分。相比之下,EquiBind直接在一步中预测精确的关键位置,而无需事先了解蛋白质的靶口袋,这被称为“盲对接”。
与大多数需要多次尝试才能在蛋白质中找到配体有利位置的模型不同,EquiBind已经具有内置的几何推理功能,可帮助模型学习分子的基本物理学,并在遇到新的,看不见的数据时成功推广以做出更好的预测。
这些发现的发布迅速引起了行业专业人士的关注,包括Relay Therapeutics的首席数据官Pat Walters。沃尔特斯建议研究小组在已经存在的用于肺癌,白血病和胃肠道肿瘤的药物和蛋白质上尝试他们的模型。虽然大多数传统的对接方法都未能成功结合在这些蛋白质上起作用的配体,但EquiBind成功了。
“EquiBind为对接问题提供了一种独特的解决方案,包括位姿预测和结合位点识别,”Walters说。“这种方法利用了来自数千种公开可用晶体结构的信息,有可能以新的方式影响该领域。
“我们感到惊讶的是,虽然所有其他方法都完全错误或只得到了一个正确方法,但EquiBind能够将其放入正确的口袋中,因此我们很高兴看到结果,”Stärk说。
虽然EquiBind已经收到了来自行业专业人士的大量反馈,帮助团队考虑了计算模型的实际用途,但Stärk希望在即将于7月举行的ICML上找到不同的观点。
“我最期待的反馈是如何进一步改进模型的建议,”他说。“我想和那些研究人员讨论...告诉他们我认为接下来的步骤是什么,并鼓励他们继续将模型用于自己的论文和自己的方法......我们已经有许多研究人员联系并询问我们是否认为该模型对他们的问题有用。
这项工作部分由药物发现和合成联盟资助;贾米尔诊所;DTRA发现针对新兴威胁的医疗对策计划;DARPA加速分子发现计划;麻省理工学院-武田奖学金;NSF探险队资助了合作研究:通过代码了解世界。
这项工作是为了纪念Octavian-Eugen Ganea,他为几何机器学习研究做出了重要贡献,并慷慨地指导了许多学生 - 一位有着谦逊灵魂的杰出学者。
免责声明:本文由用户上传,如有侵权请联系删除!
猜你喜欢
- 庆余年哪集是范闲背诗的(庆余年范闲背诗第几集简介介绍)
- 西游记里面的故事简介(西游记的故事有哪些简介介绍)
- dnf男街霸三觉(dnf86级男街霸\/千手罗汉\/暗街之王二觉刷图加点)
- 产品整体概念的主要内容是什么(什么是产品整体概念简介介绍)
- 英雄联盟手游内测怎么申请内测申请攻略(LOL英雄联盟手游内测在哪申请)
- 剑灵一个南天国金币可以换多少银币(剑灵南天国铁币,银币在哪获得)
- 凯里欧文到底多高(凯里欧文的身高体重是多少简介介绍)
- 申请工伤认定所必需的材料是什么(申请工伤认定所必需的材料是)
- 生日歌歌词(蝶变新生的主题歌歌词)
- 中餐与西餐有什么区别(中餐与西餐有什么区别)
- 索爱k506c(用索爱k510的进一下)
- 我们结婚了初恋夫妇表演舞台(我们结婚了初恋夫妇(泰民)
最新文章
- 中国好声音如果没有你李昊瀚(山野中国好声音李昊瀚唱的那么好为什么淘汰)
- 被套的尺寸是多少(被套尺寸一般是多少简介介绍)
- 怪物x联盟复刻版攻略(怪物x联盟复刻祥云马)
- 阳历是快的还是慢得(快的和慢的哪个是阳历简介介绍)
- 英雄联盟赵信特战先锋(特战先锋德邦总管赵信)
- 凤凰传奇有一首歌叫什么(凤凰传奇有一首歌歌词有)
- 为什么腾讯视频看不了直播(腾讯lpl视频看不了怎么办)
- Blue(Da(Ba Dee) 歌词)
- 联想z475开机黑屏(联想Z475开机超慢怎么回事)
- 吴建豪舞林大会跳的舞(2011舞林大会吴建豪怎么没有看见进复赛)
- 海清结婚了吗现在怎么样了(海清结婚了吗)
- 开十字绣店到哪里进货(开十字绣店在哪里进货怎么进货呢)
- 卫庄大战六剑奴是哪一集(卫庄哪集说的六剑奴是值得一战的对手)
- 微信六年来第一次开始“变脸”为什么
- iphone怎么看已连接wifi密码(iPhone怎么越狱啊)
- 求K233次列车(15车厢的座位号)
- 能链综合能源港里的充电站为何成为香饽饽
- 鸡蛋怎么做比较有营养(鸡蛋怎么做比较好吃)
- lol手游霞怎么出装(LOL新英雄霞与洛逆羽霞如何出装霞怎么出装)
- 穿越火线什么时候上架(穿越火线什么时候能玩)
- 北比臼舅怎么读(北比臼日怎么读)
- 创世之柱任务有什么用(创世之柱任务怎么做)
- 徐磊的歌曲(写给你的歌 徐磊乐演唱作品)
- 广州市经济适用住房准购证明怎么办理(如何取得广州市经济适用住房准购证明)